
“Put simply,” says AdvaMed, the trade association of the medical device industry, “the [user fee agreement recently reached between FDA and the medical technology industry] is good for FDA; it is good for industry; and most of all, it is good for American patients.”
The House version of the FDA user fee bill, which is currently being marked up, is “widely expected to contain more industry-friendly provisions, especially for medical device makers,” according to Politico (here).
“One in particular is the HELP bill’s efforts to streamline the FDA’s ability to reclassify the risk level of devices. Whether a device is deemed more, or less, risky can dramatically change the amount of clinical data and other studies required for approval.
“Currently, such reclassification is a long-term rulemaking process that must be cleared by Health and Human Services and the Office of Management and Budget along with the full complement of public hearings and comment periods. The HELP bill would turn that into a faster administrative process without the extra layers of oversight.”
According to Politico, AdvaMed is still pushing something in between “to preserve some of our due process rights,” AdvaMed’s head of government relations (ie, chief lobbyist).
Details of “FDA’s ability to reclassify the risk level of devices” may be hidden in the bills. One of these details may concern “a loophole in the law that allows [medical device manufacturers] to submit new products to the FDA for instant review as long as they classify them as an upgrade even if the product has changes that could affect safety,” says Consumer Reports. “Companies now use the process 90 percent of the time, according to a report published by Rep. Ed Markey, D-Mass., who is an advocate for industry reform.”
Meanwhile, the FDA wants to assign a new bar-code-like identification number to medical devices to help it detect malfunctions in devices AFTER they have been approved. By tapping into medical and billing records from hospitals and insurance companies, FDA hopes to identify faulty devices before they cause deaths, such as the 686 deaths from 2009 to last year connected to automated external defibrillators and at least 20 deaths recently linked to surgically-implanted heart defibrillator wires.
One of the leading manufacturers of heart defibrillation devices is Guidant. A few years ago, it had to recall one of its devices that was linked to several deaths (see NYT article). That derailed a takeover bid by Johnson and Johnson (JNJ). Meanwhile, JNJ is actively growing its medical device business and will soon acquire Synthes — a Swiss manufacturer of orthopaedic devices — for $21.3 billion. Devices now account for 40% of JNJ’s worldwide sales (see chart below; source of data: CNNMoney.com and Q1 2012 financial statement).
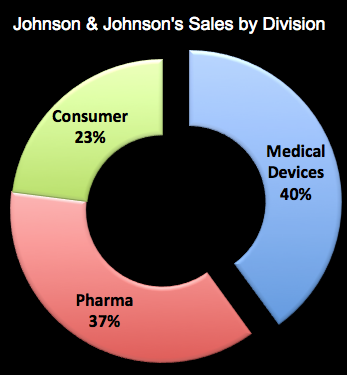
JNJ may position itself as a “consumer” products company, but its main business is pharmaceutical drugs and medical devices. With the acquisition of Synthes, which had sales of nearly $4 billion last year, JNJ’s device business will be an even bigger slice of its global sales pie (maybe 43%).











{kind=link}