Sanofi-Aventis (S-A) submitted 14 pages of comments to Docket No. FDA‐2009‐N‐0441 regarding Promotion of FDA‐Regulated Medical Products Using the Internet and Social Media Tools (find it here).
“As part of routine monitoring of online discussions hosted, or with participation, by a company, postings that contain potential adverse event reports should be addressed according to established company policies and procedures for handling and reporting spontaneous adverse event reports according to current FDA regulations,” said S-A in its comments. “This includes, when appropriate [my emphasis], to follow-up to obtain the necessary elements needed to report an adverse event.”
Considering the problems that S-A is experiencing on its VOICE Facebook page with regard to “disgruntled patient” Shirley Ledlie (see “Disgruntled Patient Shuts Down sanofi-aventis Facebook Page“), I was wondering if it is appropriate for S-A to follow up with her reports of permanent hair loss side effects of the drug Taxotere.
First, what is an adverse event and is what Shirley’s reporting an adverse event?
The definition of “adverse event” for both nonprescription drugs and dietary supplements is “any health-related event associated with the use of a [nonprescription or dietary supplement] that is adverse” (see source). This could include any unfavorable and unintended sign including an abnormal laboratory finding, symptom or disease, and, more seriously, death.
FDA defines Adverse drug experience as “Any adverse event associated with the use of a drug in humans, whether or not considered drug related, including the following: An adverse event occurring in the course of the use of a drug product in professional practice; an adverse event occurring from drug overdose whether accidental or intentional; an adverse event occurring from drug abuse; an adverse event occurring from drug withdrawal; and any failure of expected pharmacological action.” (See here)
Shirley reported hair loss, which is known side effect of Taxotere. Are known side effects, especially those listed in the drug labeling (as is hair loss for Taxotere), considered “reportable” adverse events? I found nothing from searching FDA AE reporting pages to indicate that manufacturers are NOT responsible for reporting adverse events if the event is listed as a side effect in the labeling. Besides, Shirley is reporting permanent hair loss, which is not specifically mentioned in the labeling.
In my opinion, therefore, what Shirley has reported in comments to S-A’s VOICE Facebook page is a “potential” adverse event report that requires S-A to follow-up.
Shirley’s adverse event also satisfies the FDA’ guidance on postmarketing adverse reporting, which S-A cites in its comments to FDA: “Before considering any clinical incident for submission to the FDA in an expedited or periodic safety report, applicants, manufacturers, and licensed manufacturers should have knowledge of the following four dats elements:
- An identifiable patient;
- And identifiable reporter;
- A suspect drug, biological product, or device; and
- An adverse event or fatal outcome.
S-A has all four data elements in this case. IMHO, therefore, it is appropriate for S-A not only to follow-up with Shirley, but to submit an adverse event report to the FDA. Has it done so? According to Shirley, S-A refuses to speak with her. if that is true, then S-A is not following its own recommendations it suggested in its comments to the FDA.
Does having a disclaimer about adverse events on its FB page mean that S-A does not have to report this adverse event to the FDA?
The disclaimer states: “”This page is not intended as a forum for discussing sanofi-aventis’ or other companies products including the reporting of side effects associated with the use of prescription drugs. As such, Postings that contain product discussions may be removed by sanofi-aventis… If you, or someone you know, have possibly experienced a side effect while taking any sanofi-aventis product, please contact our Drug Safety department at 1-800-633-1610, option 2, or via fax at 1-908-203-7783. You are also encouraged to report negative side effects of prescription drugs to the FDA. To do so, visit www.fda.gov/medwatch or call 1-800-FDA-1088. Postings may be reviewed by sanofi-aventis and may be subject to removal if they are deemed to be inappropriate or inconsistent with the intended use of this site. We reserve the right to block users who violate the terms of use.”
S-A suggests the same kind of wording in its comments to FDA, but does NOT say that having such a disclaimer makes it unnecessary for S-A or other drug companies from fulfilling their obligations to report AEs according to FDA law. I think the FDA would agree that it is still necessary to report adverse events found in comments submitted by an identifiable reporter.
In terms of processing AEs on company-owned social media sites, S-A says that “drug sponsors should monitor (through active participation) their own websites for potential adverse events and report according to existing internal standards. It recommends the following monitoring “parameters:”
- Development of search terms (product/disease specific) and timeframes
- Development of process for identifying and transferring potential adverse events to the company’s Pharmacovigilance department
- Training monitoring staff (in-house and/or vendor) on the aforementioned process
- Training vendors on company standard operating procedures (SOPs) for adverse event reporting
- Internal/vendor audits to assess compliance
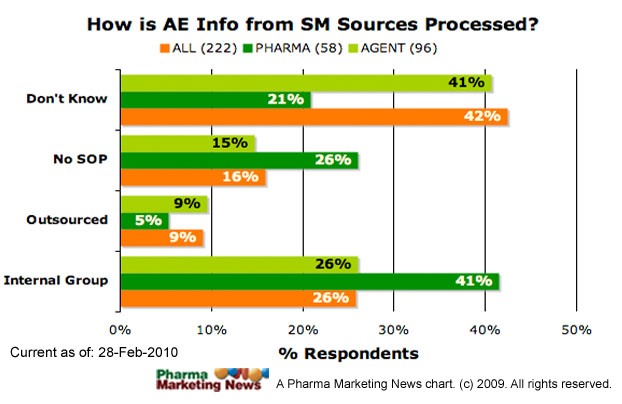
Has S-A implemented these monitoring parameters? About 26% of pharmaceutical companies may not have any social media adverse event monitoring standard operating procedures according to my recent survey (see “WANTED: Answers to FDA’s Questions Regarding Pharma’s Use of Social Media” and figure below):











{kind=link}